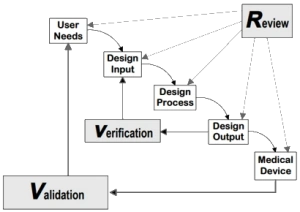
Design Transfer is a key concept in transferring the device design from development to manufacturing. But what should a person in charge of the Design Transfer focus on?
regulatory basis
The regulatory requirements for Design Transfer stem from the US FDA 21 CFR 820.30 (h). For the EU, there is no regulatory requirement in MDR 2017/745, as the Quality Management System (QMS) standard ISO 13485 does define the processes for design transfer in chapter 7.3.8.
Design Transfer is defined by the US FDA as the requirement to establish and maintain procedures to ensure that the device design is correctly translated into production specifications.
In ISO 13485, the chapter on design transfer states that there shall be document procedures for transfer of design and development outputs to manufacturing. These procedures shall ensure that design and development outputs are verified as suitable for manufacturing before becoming final production specifications and that production capability can meet product requirements.
With the definitions in the US FDA 21 CFR 820.30 and the ISO 13485, differences can be seen between the requirements for medical device Design Transfer in the USA and in the ISO 13485, which are given in the following table.
| Basis | Procedures | Design (Output) | Verification | Specification | Capability |
|---|---|---|---|---|---|
|
US FDA |
X |
X |
|
X |
|
|
ISO 13485 |
X |
X |
X |
X |
X |
Design Outputs and Verification
As each design output needs to be verified, it is common practice to check on completeness of design output verification by the help of a traceability matrix. With such a matrix each user requirement is traced to one or many design inputs, are traced to one or many design outputs and each design output shall be verified.
The use of the traceability matrix
As the traceability matrix is common practice to the device engineering department, it becomes handy for the Design Transfer. The traceability matrix, and more specifically the listed design outputs can be stated to the manufacturing engineering as the relevant outputs to consider. Ultimately, these design outputs are to be transferred into manufacturing specifications.
But, there is more to it which needs to be transferred from the design development into manufacturing. The risk management file, compiled as far as to include the hazard assessment, the use-related risk management and design risk assessments (sometimes including system risk assessments), need to be transferred as well to include the severity scale for process risk assessment and critical quality attributes.
The control strategy
Even though there is no regulatory nor ISO 13485 requirement to establish a control strategy, it has become common practice to do so. Critical attributes, risk items with a high severity, need to be controlled more stringent than other risks. Thus, the concept of critical quality attributes (CQA) can be taken to label such risks for rigorous testing and controlling.
In adoption of the concepts of ICH Q8/Q9, the control strategy can be established for the medical device during Design Transfer by the manufacturing engineer. As an outcome, the control plan is like a recipe for manufacturing on how to test what, when, and with how many samples. It is within the control plan where all the incoming inspections, in-process controls (IPC) and lot release tests are listed and justified.
The control strategy, and specifically the control plan, is the core document for manufacturing.
how to link traceability matrix and control strategy
As the traceability matrix traces user requirements to design inputs to design outputs and into verification and validation, the control plan traces CQAs to testing methods, such as IPC.
The link between these two lists are the design outputs. Design outputs are verified and traced in the traceability matrix and the same design outputs are assessed for criticality and whether they become a CQA to be traced into testing.
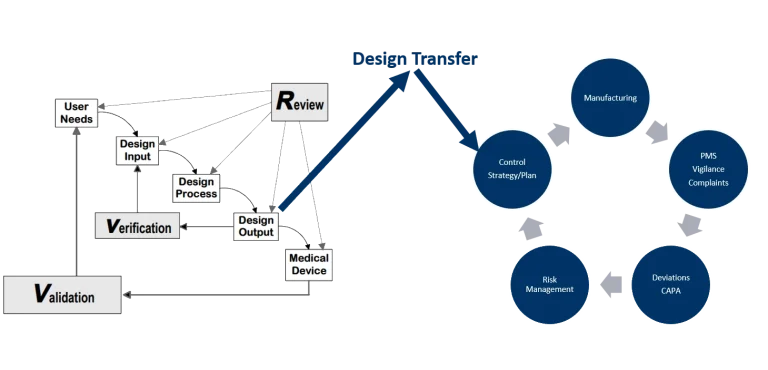
Changes, changes, changes
Keeping this link in mind, future changes are demystified and easy to conduct, even though they influence the design of the medical device.
Working backwards through the control plan during a change assessment will lead to design outputs. These same design outputs are in the traceability matrix and lead to design inputs and to verification testing. Thus, the assessment of a change is with low complexity and assessed fast.
How Avanti Europe can help
Avanti Europe’s Experts have a decade-long track record and expertise in consulting and hands-on working in process implementation in the Pharmaceutical, Cosmetical, and Medical Device industry. Our experts support your company with hands-on workforce and support in risk-based process design, documentation, and training for the company staff. Visit our online shop for checklists and other services.